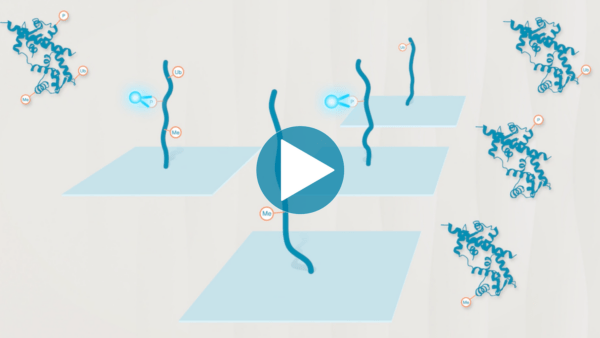
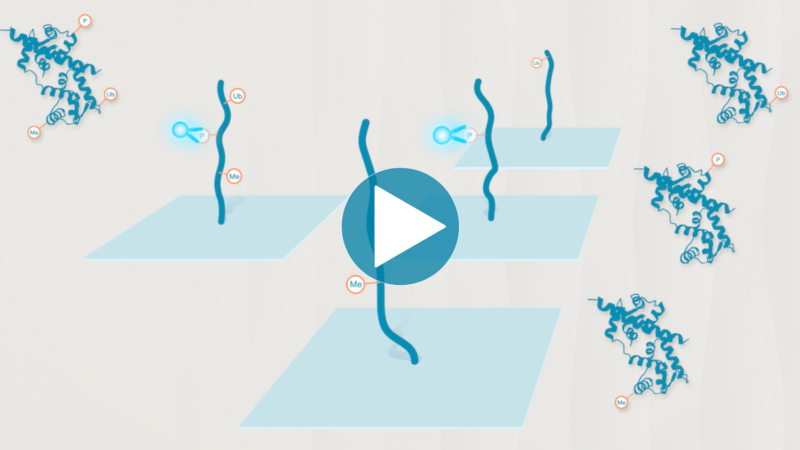
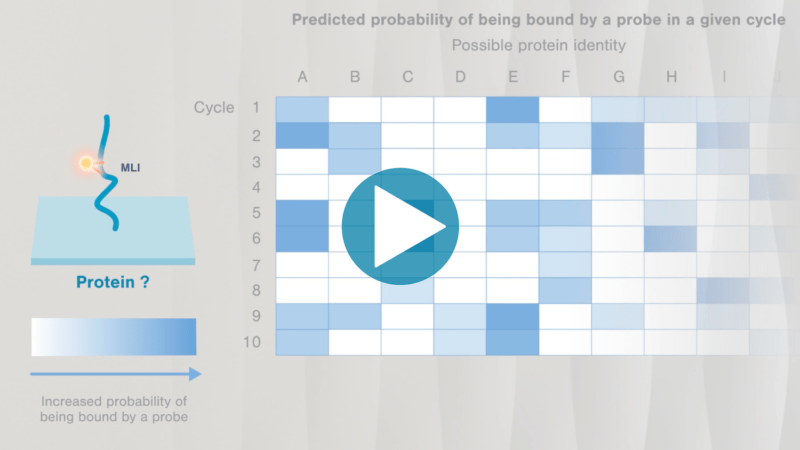
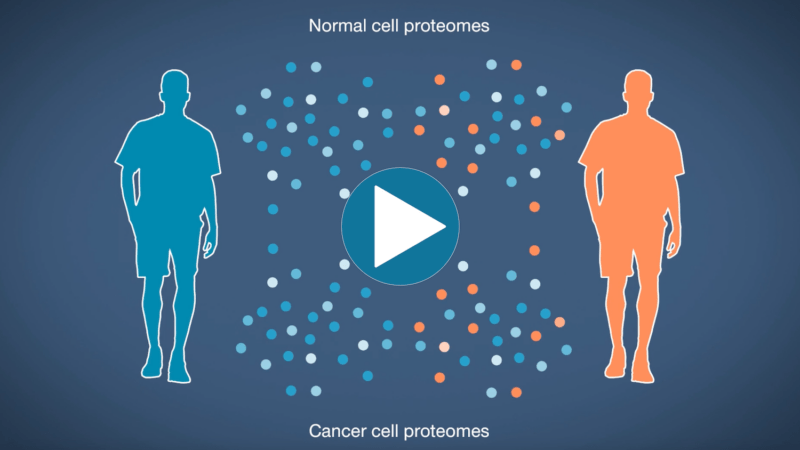
Omics-scale technologies aim to capture all the information contained in full sets of biological molecules like DNA (genomics), RNA (transcriptomics), and proteins (proteomics). Until recently, it was only possible to capture and store the data contained in the genome and transcriptome. While we have learned much about genetic diseases and biological variation from genomics and transcriptomics, DNA and RNA do not reveal the biological functions active in cells now. Proteins, on the other hand, carry out most biological functions, and proteomic technologies can provide robust analyses of active biological functions. Thankfully, we now have the technological capacity to capture, store, and analyze comprehensive proteomic data and thereby generate far deeper insights into biology than ever before. By making a proteomics platform that is accessible to all researchers, Nautilus hopes to enable researchers and physicians to apply proteomic insights to create new diagnostics, medicines, biotechnologies, and more.